Abstract
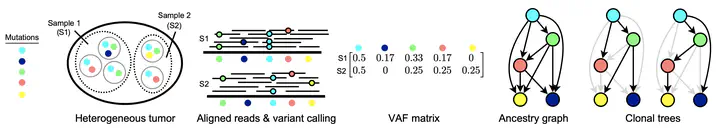
A number of methods have recently been proposed to reconstruct the evolutionary history of a tumor from noisy DNA sequencing data. We investigate when and how well these histories can be reconstructed from multi-sample bulk sequencing data when considering only single nucleotide variants (SNVs). We formalize this as the Enumeration Variant Allele Frequency Factorization Problem and provide a novel proof for an upper bound on the number of possible phylogenies consistent with a given dataset. In addition, we propose and assess two methods for increasing the robustness and performance of an existing graph based phylogenetic inference method. We apply our approaches to noisy simulated data and find that low coverage and high noise make it more difficult to identify phylogenies. We also apply our methods to both chronic lymphocytic leukemia and clear cell renal cell carcinoma datasets.
Kiran Tomlinson
PhD Candidate, Computer Science
I’m a Computer Science PhD candidate at Cornell University advised by Jon Kleinberg, researching social choice and preference learning.