pickAtom() function. This is very inefficient, since
pickAtom() takes a long time to return (it must open a file, read
from it, and perform complicated string processing). As the result, your
program takes long to run, spending most of its time on pickAtom()
calls. Furthermore, multiple calls to pickAtom() are redundant:
it suffices to call the function only once for each protein, and store the
returned coordinate matrix in a cell array. This would make your code
run several times faster. See project1.m in the
solutions for an example of how this is done.
det(U) is -1.
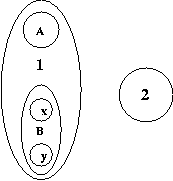
Here, clusters labeled A, x, y, and 2 are leaves, and clusters labeled 1 and B are compound. Let D(1,2) denote the distance between 1 and 2. Then, according to the project definition,
D(1,2) = (D(A,2) + D(B,2))/2 = (D(A,2) + [D(x,2) + D(y,2)]/2) / 2 = D(A,2)/2 + D(x,2)/4 + D(y,2)/4whereas, according to your implementation,
D(1,2) = D(A,2)/3 + D(x,2)/3 + D(y,2)/3The above two expressions are clearly different, and latter is incorrect, since it does not in any way acknowledge the fact that A is a leaf cluster, and x and y are clustered together.