Simple models for protein folding:
We consider the so-called H/P model on a two dimensional lattice. Ken Dill studied this system extensively as a model for protein folding. In the H/P model the amino acids are divided into two groups: Hydrophobic amino acids and Polar amino acids. Hydrophobic residues attract each other (to avoid contact with water they aggregate together). Polar residues are ambivalent and do not care with what they interact.
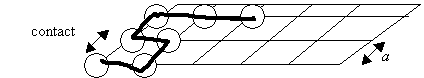
The model we considered is a cubic lattice in two dimensions.
Every amino acid occupies a point on the lattice. The minimum distance between
lattice points is ![]() and the protein chain
is self-avoiding (two amino acids cannot occupy the same position in space). It
is also the distance between two amino acids along the chain, and two amino
acids that are in contact. A contact is defined by two amino acids that are
within a distance
and the protein chain
is self-avoiding (two amino acids cannot occupy the same position in space). It
is also the distance between two amino acids along the chain, and two amino
acids that are in contact. A contact is defined by two amino acids that are
within a distance ![]() and are separated by
(at least) three bonds
and are separated by
(at least) three bonds
A protein conformation is defined as the coordinate vector
that specifies the positions of the amino acids on the lattice. The energy of a
conformation is a sum of the energies of the contacts. ![]() ,
, ![]() are the indices of the
amino acids along the chain.
are the indices of the
amino acids along the chain. ![]() is 1 if the residues
are in contact, and 0 otherwise.
is 1 if the residues
are in contact, and 0 otherwise. ![]() depends on the type
of the amino acids
depends on the type
of the amino acids ![]() . It is –1 if
. It is –1 if ![]() and
and ![]() are both hydrophobic
(both of type H), and zero otherwise (i.e. of type PP or HP).
are both hydrophobic
(both of type H), and zero otherwise (i.e. of type PP or HP).
Our goal is to find the conformation of the global energy minimum for a chain length N and a fixed sequence that includes both H-s and P-s.
First attempt: Let us think on the chain as gas (not a chain) of amino acids of type H and amino acids of type P. What is the optimal arrangement of the gas? What is the difficulty in changing back from the “gas” description to a chain?
![]()
![]()
![]()
![]()
![]() Second attempt: We are going to
optimize the energy function by a Metropolis Monte Carlo Search. A move is
defined by a single 90 degree rotation. Examples are shown below:
Second attempt: We are going to
optimize the energy function by a Metropolis Monte Carlo Search. A move is
defined by a single 90 degree rotation. Examples are shown below:


![]()
Note that the whole chain is responding to the rotation and
we must keep the chain connected. The algorithm has two random choices: (i)
pick a bond of the N-1 bonds at random; (ii) attempt a rotation to the right or
to the left at random. The energy of the new conformation is then evaluated and
the move is accepted or rejected using the Metropolis criterion for “before”
and “after” the step (i.e. ![]()
![]() ).
).
If two amino acids occupy the same position in space the new energy is set to infinite and that step is always rejected.
We start at high temperature ![]() and decrease the
temperature linearly as a function of the number of accepted steps.
and decrease the
temperature linearly as a function of the number of accepted steps.
It is possible to adjust the algorithm to obtain acceptance ratio of about 30 percent at higher temperature. For example, the bonds at the beginning and at the end of the chain are more likely to produce acceptable steps. We can therefore modify the probability of sampling a rotation of a specific bond to better control the acceptance probability. For example, we may wish to reduce the acceptance probability. We can do that by decreasing the probability of sampling a rotation at the first or at the last bond to only 10 percent. To conserve probability we should increase the sampling probability of other bonds. This choice will also depend if we are in an open or compact configuration (how?)
Our goal is to find low energy minima (correct protein folds), as we approach the zero temperature.
What is the optimal conformation of an all H polymer, of an all P polymer? Knowledge on exact solutions of some sequences can help debug the code…
There is a choice of moving the chain before the rotating bond or moving the chain after, what is the simplest choice?