




|
|
|
|
|
|
| Swamp_Slime | Shy_Frecklepuss | Fuzzy_Trible | Gray_Floop | Ballards_Protoduck |
A few of the 40 animals in the CS2110 bestiary. Credit for the pictures goes to our talented artistic consultant, Ursula Hilsdorf.
Note: This page gives an overview of the entire 5-part assignment. Below you'll find links that will take you to the detailed versions of each of the five assignments. You'll use CMS to hand in your solutions.
Link for Assignment one: Due Sept. 9 at 11:59pm
Link for Assignment two: Due Sept 22 at 11:59pm
Link for Assignment three: Due Oct 9 at 11:59pm
Link for Assignment four, Due Nov 8 at 11:59pm
Link for Assignment five, Due Dec 3 at 11:59pm. Whew!
Creating an Evolutionary Tree
During fall of 2009,
CS2110 students will be working on a semester-long project that has a series of
intermediate deliverables. Even
before explaining what this project is about we want to reiterate that
for
assignment 1, each
student is expected to work on his or her own, and for other assignments you can
work in teams of 2, but no more, and teams can't collaborate with other teams!
You are welcome to have non-specific
dialog about the project with friends or a study group, but every line of code
you type needs to be written by you personally, or by you and your teammate on
A2-A5, and you can’t show other people
your solutions before the deadline for handing them in.
(Suggestion: read the section on “automated cheating detection.)
What You’ll Do
The goal of the
project is to use genome data to figure out the relationship between a set of
animals. We’ll provide
pictures of the animals and some other
data, and as part of that data we'll give you the
animal's DNA. Using code we develop
during the semester, we’ll tackle problems such as extracting genes from the
DNA, comparing to see how similar two genes are, and finally building a graph (a
tree) representing the evolutionary history of the creatures and displaying the
data in web pages. The problem is a
simplification of one of the most important problems seen in modern biology.
Genomic computations
in “reality” can be fairly messy, so for CS2110 we’ll be using a somewhat
simplified genetic model. The simpler
version isn’t completely realistic, but it does parallel the actual
computational biology problem, while limiting itself to technical ideas that are
at the right level for students taking a second semester Java course.
We’re hoping that you’ll discover that without learning a huge amount of
computer science, you can already do some really amazing things.
We also want to see you get experience writing high-quality code – this
is a skill you can only learn by doing.
Our semester-long
project will be broken into five separate sub-projects, each linked to material
we’ll be learning in lecture.
Each subproject will involve implementing some set of Java classes that supports
a specific interface that we’ll define carefully.
(One warning:
Even if you
think the interface we request is lame, you still need to use it because our
automated testing program checks for it!)
When practical, we’ll provide a very limited test setup that you can use to make sure
that your solution does match what we expect, but then when you hand in your
solution, we’ll test it against a harder set of scenarios.
So your job is to write code that definitely works in our simple tests,
but in fact that can also work for cases that it hasn’t previously
“encountered”.
To get full credit,
each assignment that you hand in needs to be well-written, properly debugged
code that correctly implements the functionality and interface we assigned, and
needs to run on our full range of automated test scenarios. Your code has to be
properly commented and clear, with good choices of variable names and if
something can be done in a simple way, or in a complicated way, our graders will
deduct points for overly complex solutions.
(The famous phrase is KISS: Keep
it
Simple,
Stupid!)
All our projects have reasonably simple solutions.
As a safety net,
after each subproject is handed in, we’ll release Professor Birman’s solution
for that same problem. This way,
you can tackle the next subproject even if yours had serious bugs. Just the
same, we really would encourage you to work with your own code.
It will not be possible to get top grades in CS2110 unless you use your
own code entirely, or at least as much as possible.
The Project
As you know from
your biology classes, each species is defined by a unique genome: a sequence of
amino acids from a small alphabet.
The real genome is composed of triples (so-called “codons”) of letters
representing the associated amino acids: A, C, U and G.
In CS2110, we won’t worry about codons, and will work with a fake genomic
alphabet with amino-acid symbols A, C, I and O.
The genome mixes
junk (“non-coding”) DNA) with genes
that encode the proteins that make up the animals and control them.
Our CS2110 DNA is like this too.
In our DNA sequences, a gene always starts with a special three-letter
code: AOC, and ends with a second three-letter code: ICA.
Thus, DNA sequence AOACOAOCOICCACOIIICOCIOICAIIOO actually consists of a
single gene and some junk: AOACO
start
OICCACOIIICOCIO
end IIOO. The AOACO
and IIOO parts are examples of junk DNA, whereas
OICCACOIIICOCIO is an example of a gene.
Notice that the start and end sequence aren’t included in the gene itself
– they are like another form of junk.
Human genes are
billions of amino acids long.
CS2110 genes are a little shorter: a typical DNA sequence will be about 50,000
letters in length and might contain as few as 10 or as many as 50 genes, each of which would be 150
to 500 letters long. Note:
the longest DNA sequence in our experiments will never be more than
99,999 letters long. The longest
gene will never be more than 999 letters long.
When a species
reproduces, its genes change in many ways.
Even within a single species there can be small DNA differences in the
genes carried by individuals, and each child inherts some genes from each of its
parents. But a new species can
arise when gene copying mistakes occur during cellular reproduction.
For CS2110, we’ll focus on genes that arise by
duplication (e.g. an entire gene or
set of genes is simply copied twice from the parent to child),
swaps (some short sequence of letters
is swapped with some adjacent sequence),
insertions (of small numbers of letters) or
deletions.
For example, let’s
look a closer look at our sample gene from above, call it G1 :
"OICCACOIIICOCIO”.
(We underlined two letters to draw
your attention to them).
Now suppose that we were shown a gene G2 :
"OICCCAOIIICOCIO”.
A possible relationship is that G2 could have originally been the
same as G1, but then a two-letter swap occurred (AC became CA).
Of course there are also other ways this
could have happened. For example,
perhaps the A was deleted in the first gene to create a gene G’, and then later
an A was inserted right after the C to create G2.
In fact all sorts of crazy things could have happened… with these simple
rules there are infinitely many ways that G1 could have become G2.
The swap shown is just the most likely explanation – the shortest
explanation.
For CS2110, we’ll be
developing a gene comparison procedure that wouldn’t work for humans (where gene
comparison is a very complex question), but does work for our « fake » genomic
data because the genes were actually created synthetically and designed to be
relatively easy to compare. We’ll
be using a method inspired by a real gene comparison algorithms (examples of
famous ones are Smith-Wasserman and BLAST – look them up in Wikipedia if you get
interested in the topic).
The basic idea is to
define a distance metric with which we can score
gene similarity.
To make things simple, isuppose that when two genes are directly related, the
changes all occur at one part of the sequence.
Thus if G1 evolved into G2, we can rewrite G1 =
abc
and G2 = adc,
and all the changes will have occurred in the region that used to contain
b
and now contains d.
Given G1 =
abcand G2 =
adc,
such that G1 comes before G2 under the rules defined in
assignment two, we’ll define a distance score this way:
1.
Count all
the letters in
b
and all the letters in d.
For example, perhaps
b
contains 3 A’s, 2 C’s, 2 I’s and 5 O’s.
Now do the same for d:
maybe you’ll find 0 A’s, 1 C’s, 5 I’s and 9 O’s.
2.
Sum up
letters in both strings: 1 C, 2 I’s and 5 O’s for a total of 8.
We’ll score these as 15 points each: 120 points.
3. Next compute the letters in one string but not the other. We’ll score these as 8 points if the characters are found in both strings, but the number of instances differs (DCost), and 25 points if one string contains a character that the other string doesn't contain at all (DDCost). In our example: both strings contain the characters C and I and O, so we get (1+3+4)*DCost = 64 but only one string contains A's: 3*DDCost = 75, giving a total of 139.
Of course we oversimplified: changes could occur at a few places, not necessarily all in one region. Our "real" solution needs to take this into account. Given two genes, we'll start by computing the length of any prefix they have in common, e.g. XYZ <stuff> and XYZ <stuff'>. Next, we'll look for suffixes they have in common: <stuff> XYZ and <stuff'> XYZ. So now we're down to <stuff> and <stuff'>, which (if they aren't null strings) differ at least in the first and last characters, but could contain a "section" that matches.
Let's assume that if <stuff> and <stuff'> are of different lengths, then <stuff> is the shorter of the two. We'll take a "window" of MinMatch characters (4, as defined below) within <stuff> and "slide" it along <stuff'> looking for a match. For example, if <stuff> was ACIOCAO and <stuff'> was CIAACIOCCC, then we can slide ACIO (the first 4 characters of <stuff>) along <stuff'>: first we would check it against CIAA, then IAAC, then AACI and then (horray!) ACIO. Assignment two explains exactly how this needs to be done; use the detailed explanation in the assignment when coding a solution.
If we find a match, we can "grow" that match one character at a time towards the right: the longest match possible is ACIOC. So now we know that <stuff> should really be fragmented into three parts: a null string, then ACIOC, then AO. And <stuff'> into three parts too: CIA, then the matching section, ACIOC, and then the remainder, CC. We use our difference computation on the first pieces: a null string versus CIA. (Score 3*DDCost = 75) We have an exact match for ACIOC. And then we repeat this whole procedure for the remainder: <stuff> will now be AO and <stuff'> will be CC. These are shorter than MinMatch, so we're finished and we compute the difference computation on AO and CC (score 4*DDCost = 100), adding that to the running total. Our two genes are at distance 175 from each other.
Notice that had we not used this fancier scheme, we would have gotten a completely different answer and, worse, that answer would treat a matching section (ACIOC) as if it differed between the two.
Here are some constants we'll use for the sorts of comparisons just described (and for some additional ones mentioned below):
public
class Constants {This scoring
function is what we would call a “heuristic” – a rule of thumb that will suffice
for our purposes in CS2110.
It may seem a bit ad-hoc, but in fact the Smith-Wasserman and BLAST algorithms
also use heuristics, and of a very similar nature.
The problem is that computing the exact way that G1 evolved into G2 is
such a hard problem – there are too many possibilities to explore, and in
general, more than one might lead to the right place.
Worse, we can’t really know that evolution followed the shortest path.
So this is a case in which there isn’t any way to know the “true” answer.
A good rule of thumb may be about as good as we can do!
You will find that
when genes are reasonably similar, the scoring function tends to be 750 or less
(often much less). When genes are
very different the score rises sharply.
What about comparing
entire animals? We can do this using a
second kind of scoring function to compute an animal’s closest relative.
For example, perhaps cats have 73 genes in their genome and dogs have 81,
and out of these, 45 are common to the two.
We’ll use our gene comparison functions to try and measure the distance
from dogs to cats. If the two are
very closely related we would expect that all the different genes are actually
pretty closely related. In
contrast, if we find huge differences, that would suggest that dogs and cats are
pretty distant from each other in an evolutionary sense.
Of course, keep in mind that our little zoo is just a sample of all the
animals that ever existed – dogs and cats could be pretty closely related, but
there could be many other animals between their common ancestor and each
species. So here’s the scoring
function we’ll use:
Given animal A with genes {A1, …. Ak} and animal B with genes {B1, … Bn}
For each gene in A, find the closest gene in B. Add the distance to score. [Note: HW4 will slightly modify this rule]
For each gene in B, find the closest gene in A. Add the distance to score. [Note: HW4 will slightly modify this rule]
Divide score by two; this is the "animal to animal" distance.
This can be used to compute a matrix of Animal-to-Animal "distances". Given that matrix, we can now compute what is called a Phylogeny tree: we take an animal that is believed to be the most primitive ancestor in the group, and then look for an animal in our bestiary that is as similar as we can find to the root animal. We attach this best matching animal below our root. Now we repeat the process, looking for the animal that represents the closest match to any animal already in the tree, and again attaching it as a child of the animal with which the match was closest. And we continue until we've put every animal below its best matching relative. (Notice that you end up with a very different tree if you start with a different first choice -- the "root" matters!)
Each CS2110 project will come with its own assignment handout, but here’s a high-level overview of the projects and a short description of what each one entails.
Project 1:
We’ll start by writing Java classes to just read in the basic genomic data
for our menagerie of animals. Each animal will be defined by a
file that
contains the animal’s English-language name, its Latin species name, a filename where a
picture of the
animal can be found and its DNA sequence.
Most of these DNA sequences are long and we do not recommend printing the
full set of files. However, we will
have some « fake » animals with very short DNA sequences that you can work with
for testing, and those can be printed if necessary.
The files to read will be provided as arguments to your program.
As you
read each file, you’ll « parse » the contents to break out the different pieces
of information contained within it. You’ll use this to construct an object representing each animal.
Rather
than storing the entire DNA string, which may have a lot of junk, you’ll break
the DNA string up into its constituent set of genes.
Store each gene into a special Java structure called a Hashmap that we’ll learn about in class, using the gene string itself as a hash key. This will let you create a set of the entire set of genes found in our entire menagerie of animals. Assign each unique gene a number and use these to generate gene names: G1, G2, etc. The idea is this: if the program sees a new gene (one not already in the gene map) it assigns it a new name like the ones above. If an animal has a gene that has shown up previously, we’ll use the same name for that gene. Thus each separate gene name corresponds to a genetic code that differs from every previously known gene code.
We’ll
modify our project 1 code so that the gene numbers actually respect a sorting
order. This order will list shorter
genes before longer ones, and break ties alphabetically.
Thus, G1 would necessarily either be shorter than G2, or be the same
length but alphabetically smaller than G2.
We’ll also
start to work on a gene comparison algorithm that computes an estimate of the evolutionary distance between two
genes. Doing a good job of designing
the gene comparison class is important, so we’ll do this carefully, providing a
number of methods that you can use later in subsequent stages of the project.
We’ll use the gene comparison class and
will use it to compute an all-to-all matrix of relationships between all the
genes in our entire data set (computed in project 1).
This will let us list, for each gene, the gene other than itself that has
the most similarity. For example,
G1 is identical to itself, but perhaps is very different from G2.. G47, rather
similar to G48, etc. So G1 might be
“most similar” to G48. G48, on the
other hand, might be “most similar” to G19, etc.
First we'll enhance the gene-to-gene comparison table from A2 with a slightly fancier comparison policy that gives more accurate results. This is explained more carefully in the handout.
Next build a second all-to-all matrix giving animal-to-animal comparison scores, using the method described earlier, for each pair of animals. Our scheme will have the property that A compared to B and B compared to A give the same score.
Write a
method that, for each animal, looks up its closest relative.
Using a Java display interface that
we'll explain how to build in the detailed handout, build an interactive user interface that shows animals and
their pictures and, when an animal is clicked, highlights the animals most
similar to it. It will do this by
redrawing the background color of the selected animal’s picture in a bright color
and then recoloring all the other animals with colors that vary based on
their distance from the selected one.
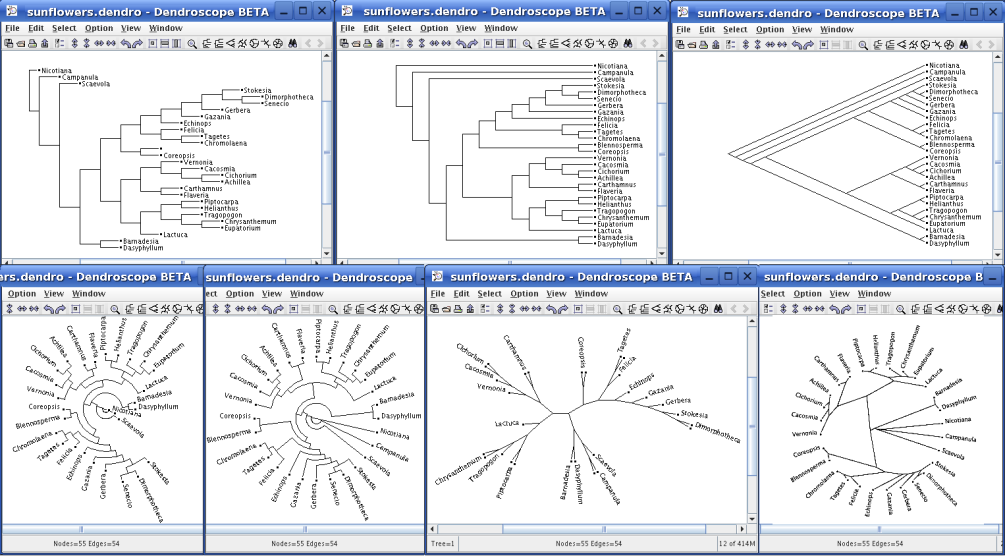
A typical input file
for Dendroscope would have a name like “Beasts.txt” and would contain a line
like this:
(A1:0.0097,(A2:0.45,A3:0.717)).
You just tell the program to read
in the file from the file « open » command and it generates the tree
automatically. Initially, no
animals will be shown, but if you use the « option » that tells the program
where to find the image files, it will display their pictures too.
You can also put this information right inside the input file if you
like. Obviously, you’ll need to
produce this file from your program, not by hand.
If the input file
uses node names like we did above (A1, A2, A3), then
Dendroscope would expect that pictures of the animals are in files named
A1.bmp, A2.bmp, etc. Of
course A1 is just a name and you can use anything else : Zebra would work, or
Freckleopolous, etc.
The floating point
numbers tell Dendroscope how long to make the edges in the graph – if you don’t
give a number, it uses 1.0 as the default.
Using .0097 for A1 causes it to be displayed « high » in the tree
relative to A2 and A3 ; A2 will be below A1 and A3 end up lowest of all.
The program offers many tree representations and you can try different
versions, depending on your mood.
We’ve noticed that
some Cornell classes do a so-so job of enforcing the academic integrity policy.
In CS2110, we have a solution: we’re using an automated system that uses
sophisticated artificial intelligence techniques combined with some pretty fancy
program analysis tools to notice unusual similarities between programs turned in
by different people. It is
important to realize that these tools really work and that they are quite hard
to fool. So while it might seem
tempting to borrow a solution from some, change the variable names and comments,
or reorder the statements, our tools would be very likely to figure out what you
did. We take cheating
seriously, and cheating with an attempt to cover it up is grounds for failing
the course outright. Realistically,
it is much easier to just do the assigned problems than to get away with handing
in code someone else wrote, because short of rewriting that code completely from
scratch, we’ll catch it.
So you’ve been
warned:
It is just not possible to get away with
cheating in CS2110. Please do
your own work, and come talk to Professor Birman, or the TAs, as often as needed
if you get stuck and need help.
We’ll get you back on track. In
contrast, borrowing a solution from a pal will just get both of you into very
serious trouble.